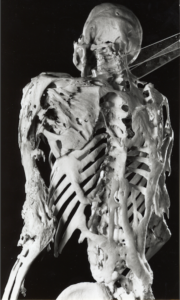
Fibrodysplasia ossificans progressiva is an incredibly rare disease, striking just one out of every two million people. It’s also an incredibly astonishing disease. A single mutation to a single gene causes muscles to spontaneously turn into new bones. Over time, people with fibrodysplasia ossificans progressiva (FOP for short) grow a second skeleton–one that can cut their lives short.
I wrote about FOP in “The Girl Who Turned to Bone” in the Atlantic in 2013. At the time, FOP served as a microcosm for the struggles of people with rare diseases. (In the United States, almost 30 million people have rare diseases of one kind or another.)
Rare diseases have historically attracted little interest from scientists or pharmaceutical companies. Working on common diseases like cancer or diabetes seemed more likely to lead to rewards, both academic and financial. As I wrote in my article, Fred Kaplan of the University of Pennsylvania got a lot of puzzled looks from his colleagues when he decided in the late 1980s to dedicate his career to figuring out FOP. It seemed like professional suicide. And he has certainly traveled a hard road since then. It took many years for him and his colleagues to find the gene behind FOP. And then they spent many more years trying to understand how a mutation to that gene actually leads to the disease. In the meantime, Kaplan has not had any effective treatment to offer his patients with FOP.
I ended my Atlantic story on a hopeful note, observing how rare diseases like FOP were starting to gain more attention–thanks in part to the efforts of patients themselves, as well as new initiatives from the National Institutes of Health. But in the two years since the story came out, things have changed a lot faster than I would have predicted. Rare diseases are attracting a huge amount of attention, which is leading to some potential treatments. One sign of this progress is a study on FOP that’s being published today in the journal Science Translational Medicine. A team of scientists has found a molecule that appears to block the second skeleton.
The gene behind the disease, called ACVR1, encodes a molecule that sits on the surface of cells. There it can grab signaling molecules and relay messages to the interior of the cell. A team of scientists at Regeneron Pharmaceuticals engineered human cells so that they carried the mutation to ACVR1 that is found in people with FOP. Then they hurled a barrage of molecules at the cells, to see if the mutant ACVR1 responded in a peculiar way to any of them. They discovered one that triggered just such an odd response.
The molecule is called activin A. It is released by cells to help with many different tasks in the body, from the development of organs in embryos to healing wounds. The fact that activin A helps heal wounds is especially intriguing when you consider one of the most striking features of FOP: people with the disease often abruptly grow new bones after injuries–even slight ones like bumping into a table corner. Kaplan and other researchers have long wondered if the mutation that causes FOP alters the body’s ability to heal wounds. Instead of causing stem cells to rebuild muscle and other damaged tissue, the body might signal them to become bones. And activin A might carry that faulty signal. In normal cells, it silences ACVR1, but in mutant cells, it excites the receptor.
To test that possibility, the scientists developed mice that carry the FOP-causing mutation. The mice formed new bones in much the same way people with FOP do. The scientists found that if they inserted a sponge soaked in activin A into the mice, the sponge turned to bone.
If they could block activin A, the scientists reasoned, they might be able to stop the chain reaction that creates new bones. In another line of research, Regeneron scientists had developed antibodies that latched onto activin A–and only activin A. When the scientists injected these antibodies into FOP-model mice, they prevented the animals from forming any new bones. Full stop.
The fact that researchers at a company like Regeneron made this discovery is telling. Pharmaceutical companies have increasingly turned their attention to rare diseases in recent years, because, paradoxically, the rare disease market may turn out to be very profitable.
Companies are rolling out drugs with sky-high price tags. Even if they’re used by relatively few people, the companies can make a lot of money. In one demonstration of how times have changed, the rare-disease company called Baxaltra is going to be bought soon for a reported $30 billion. Whether the expensive price for rare-disease drugs are really justified, however, is becoming a matter of intense debate.
Fred Kaplan, who wasn’t involved in the new study, hailed it as a “landmark” in a commentary he wrote with colleagues for the International Fibrodysplasia Ossificans Progressiva Assocation. But there are still many challenges scientists will have to face before activin A can become an effective drug for FOP.
For one thing, researchers have to see if it works as well in people as it does in mice. But because people with FOP are so sensitive to injuries (even a muscle injection can trigger a new bone), regular human trials won’t work. Fortunately, Kaplan and his colleagues have discovered that they can harvest bone-generating stem cells out of baby teeth from children with FOP. So they’re now trying to replicate the activin A studies with these cells.
Setting aside the possible medical potential of this research, it drives home just how mysterious rare diseases can be. FOP might seem like it should be a simple disease to treat. After all, it’s caused by a single mutation to a single gene. But it’s actually fiendishly complex, because it disturbs an intricate web of chemical reactions that our bodies use to grow muscles and bones. The search for a cure for FOP has been going on for over a quarter of a century, and yet no one thought to consider activin A. A normal version of ACVR1 doesn’t relay activin A’s messages. And so no one even guessed that a mutant version would.
“As Alfred Hitchcock demonstrated with clarity,” Kaplan and his colleagues write, “the best way to conceal reality is to hide critical clues in broad daylight.”
[Update: Corrected the affiliation of the researchers]
Copyright Carl Zimmer 2015.